Multiome Workflow¶
Below is the workflow for SCRIPro’s multi-omics data input, which requires paired/unpaired scRNA-seq and scATAC-seq data as input.
To demonstrate SCRIP’s ability to be applied to Multi-omic data, we applied SCRIP to 10X Multiome-seq lymphoma sequencing data.Data are available on https://www.10xgenomics.com/datasets/fresh-frozen-lymph-node-with-b-cell-lymphoma-14-k-sorted-nuclei-1-standard-2-0 .
Using Shell:¶
Transcription factor enrichment scores can be obtained by SCRIPro using the following shell statement:
scripro enrich_multiome -i ./data/rna/rna.h5ad -n 50 -s hg38 -a matrix -b 0 -f ./data/atac/atac.h5ad -g./gencode.v43.chr_patch_hapl_scaff.annotation.gtf.gz -p multiome -t 12
Using Python for custom analysis:¶
import anndata as ad
import networkx as nx
import scanpy as sc
import scglue
from matplotlib import rcParams
import numpy as np
import scripro
import pandas as pd
import warnings
import seaborn as sns
from itertools import chain
from scipy.spatial.distance import cdist
warnings.filterwarnings("ignore")
Load data¶
Respectively loaded ATAC-seq and RNA-seq data.
scglue.plot.set_publication_params()
rcParams["figure.figsize"] = (4, 4)
rna = sc.read_h5ad("/fs/home/xuyunfan/data/10x/lymph2/rna/rna.h5ad")
rna.var_names_make_unique()
rna.raw = rna
atac = sc.read_h5ad('/fs/home/xuyunfan/data/10x/lymph2/atac/atac.h5ad')
Use GLUE to compute scATAC-matched scRNA barcodes¶
Here, we use GLUE to align the two omics data,here is the normal GLUE workflow:
atac.var_names_make_unique()
rna.layers["counts"] = rna.X.copy()
sc.pp.normalize_total(rna, target_sum=1e4)
sc.pp.log1p(rna)
sc.pp.highly_variable_genes(rna, min_mean=0.0125, max_mean=3, min_disp=0.5)
sc.pp.scale(rna)
sc.tl.pca(rna, n_comps=100, svd_solver="auto")
sc.pp.neighbors(rna, metric="cosine")
sc.tl.umap(rna)
sc.pl.umap(rna)

scglue.data.lsi(atac, n_components=5, n_iter=15)
sc.pp.neighbors(atac, use_rep="X_lsi", metric="cosine")
sc.tl.umap(atac)
sc.pl.umap(atac)

scglue.data.get_gene_annotation(
rna, gtf="../data/gencode.v43.chr_patch_hapl_scaff.annotation.gtf.gz",
gtf_by="gene_name"
)
rna.var.loc[:, ["chrom", "chromStart", "chromEnd"]]
| chrom | chromStart | chromEnd | |
|---|---|---|---|
| MIR1302-2HG | chr1 | 29553.0 | 31109.0 |
| FAM138A | chr1 | 34553.0 | 36081.0 |
| OR4F5 | chr1 | 65418.0 | 71585.0 |
| AL627309.1 | NaN | NaN | NaN |
| AL627309.3 | NaN | NaN | NaN |
| ... | ... | ... | ... |
| AC141272.1 | NaN | NaN | NaN |
| AC023491.2 | NaN | NaN | NaN |
| AC007325.1 | NaN | NaN | NaN |
| AC007325.4 | NaN | NaN | NaN |
| AC007325.2 | NaN | NaN | NaN |
36621 rows × 3 columns
genes_to_remove = rna.var[~(rna.var.loc[:,"chromStart"]>0)].index
rna = rna[:, ~rna.var.index.isin(genes_to_remove)]
atac.var_names[:5]
Index(['chr1_9795_10696', 'chr1_17061_17939', 'chr1_180997_181703',
'chr1_183968_184757', 'chr1_186502_187406'],
dtype='object')
split = atac.var_names.str.split(r"[_]")
atac.var["chrom"] = split.map(lambda x: x[0])
atac.var["chromStart"] = split.map(lambda x: x[1]).astype(int)
atac.var["chromEnd"] = split.map(lambda x: x[2]).astype(int)
atac.var.head()
| chrom | chromStart | chromEnd | |
|---|---|---|---|
| chr1_9795_10696 | chr1 | 9795 | 10696 |
| chr1_17061_17939 | chr1 | 17061 | 17939 |
| chr1_180997_181703 | chr1 | 180997 | 181703 |
| chr1_183968_184757 | chr1 | 183968 | 184757 |
| chr1_186502_187406 | chr1 | 186502 | 187406 |
rna.var
| highly_variable | means | dispersions | dispersions_norm | mean | std | chrom | chromStart | chromEnd | name | ... | itemRgb | blockCount | blockSizes | blockStarts | gene_id | gene_type | tag | hgnc_id | havana_gene | artif_dupl | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MIR1302-2HG | False | 1.000000e-12 | NaN | 0.000000 | 0.000000 | 1.000000 | chr1 | 29553.0 | 31109.0 | MIR1302-2HG | ... | . | . | . | . | ENSG00000243485.5 | lncRNA | ncRNA_host | HGNC:52482 | OTTHUMG00000000959.2 | NaN |
| FAM138A | False | 1.000000e-12 | NaN | 0.000000 | 0.000000 | 1.000000 | chr1 | 34553.0 | 36081.0 | FAM138A | ... | . | . | . | . | ENSG00000237613.2 | lncRNA | NaN | HGNC:32334 | OTTHUMG00000000960.1 | NaN |
| OR4F5 | False | 5.497313e-03 | 1.040101 | -1.097506 | 0.002889 | 0.056353 | chr1 | 65418.0 | 71585.0 | OR4F5 | ... | . | . | . | . | ENSG00000186092.7 | protein_coding | NaN | HGNC:14825 | OTTHUMG00000001094.4 | NaN |
| OR4F29 | False | 1.000000e-12 | NaN | 0.000000 | 0.000000 | 1.000000 | chr1 | 450739.0 | 451678.0 | OR4F29 | ... | . | . | . | . | ENSG00000284733.2 | protein_coding | NaN | HGNC:31275 | OTTHUMG00000002860.3 | NaN |
| OR4F16 | False | 1.000000e-12 | NaN | 0.000000 | 0.000000 | 1.000000 | chr1 | 685715.0 | 686654.0 | OR4F16 | ... | . | . | . | . | ENSG00000284662.2 | protein_coding | NaN | HGNC:15079 | OTTHUMG00000002581.3 | NaN |
| ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... | ... |
| MT-ND4 | True | 2.037123e+00 | 4.331923 | 3.915107 | 0.950185 | 1.276617 | chrM | 10759.0 | 12137.0 | MT-ND4 | ... | . | . | . | . | ENSG00000198886.2 | protein_coding | NaN | HGNC:7459 | NaN | NaN |
| MT-ND5 | True | 6.776105e-01 | 3.704260 | 5.513758 | 0.219016 | 0.636512 | chrM | 12336.0 | 14148.0 | MT-ND5 | ... | . | . | . | . | ENSG00000198786.2 | protein_coding | NaN | HGNC:7461 | NaN | NaN |
| MT-ND6 | True | 2.098734e-01 | 3.157219 | 1.393144 | 0.062790 | 0.337936 | chrM | 14148.0 | 14673.0 | MT-ND6 | ... | . | . | . | . | ENSG00000198695.2 | protein_coding | NaN | HGNC:7462 | NaN | NaN |
| MT-CYB | True | 1.438881e+00 | 4.125400 | 4.742718 | 0.554779 | 1.015753 | chrM | 14746.0 | 15887.0 | MT-CYB | ... | . | . | . | . | ENSG00000198727.2 | protein_coding | NaN | HGNC:7427 | NaN | NaN |
| MAFIP | False | 2.067433e-02 | 1.815702 | -0.185062 | 0.008163 | 0.110646 | GL000194.1 | 53593.0 | 115055.0 | MAFIP | ... | . | . | . | . | ENSG00000274847.1 | protein_coding | NaN | HGNC:31102 | NaN | NaN |
23469 rows × 24 columns
guidance = scglue.genomics.rna_anchored_guidance_graph(rna, atac)
guidance
scglue.graph.check_graph(guidance, [rna, atac])
scglue.models.configure_dataset(
rna, "NB", use_highly_variable=True,
use_layer="counts", use_rep="X_pca"
)
scglue.models.configure_dataset(
atac, "NB", use_highly_variable=True,
use_rep="X_lsi"
)
guidance_hvf = guidance.subgraph(chain(
rna.var.query("highly_variable").index,
atac.var.query("highly_variable").index
)).copy()
glue = scglue.models.fit_SCGLUE(
{"rna": rna, "atac": atac}, guidance_hvf,
fit_kws={"directory": "glue"}
)
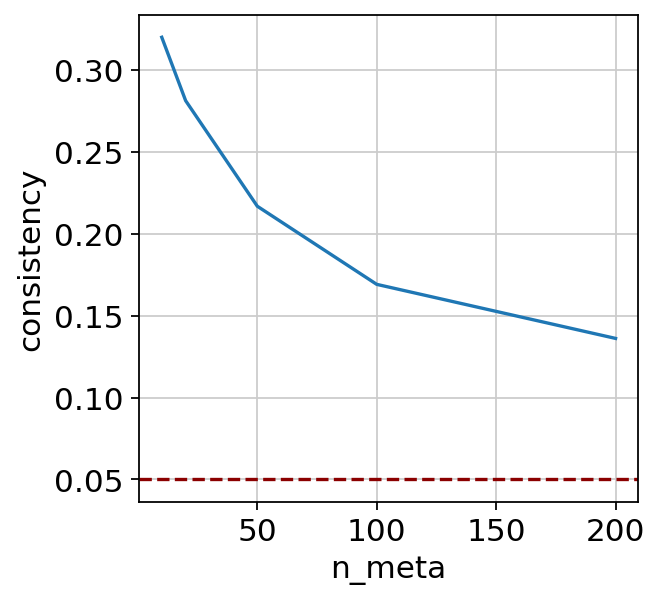
dx = scglue.models.integration_consistency(
glue, {"rna": rna, "atac": atac}, guidance_hvf
)
dx
| n_meta | consistency | |
|---|---|---|
| 0 | 10 | 0.320081 |
| 1 | 20 | 0.281343 |
| 2 | 50 | 0.216881 |
| 3 | 100 | 0.169161 |
| 4 | 200 | 0.136142 |
_ = sns.lineplot(x="n_meta", y="consistency", data=dx).axhline(y=0.05, c="darkred", ls="--")
rna.obsm["X_glue"] = glue.encode_data("rna", rna)
atac.obsm["X_glue"] = glue.encode_data("atac", atac)
rna
AnnData object with n_obs × n_vars = 14566 × 23469
obs: 'balancing_weight'
var: 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'mean', 'std', 'chrom', 'chromStart', 'chromEnd', 'name', 'score', 'strand', 'thickStart', 'thickEnd', 'itemRgb', 'blockCount', 'blockSizes', 'blockStarts', 'gene_id', 'gene_type', 'tag', 'hgnc_id', 'havana_gene', 'artif_dupl'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', '__scglue__'
obsm: 'X_pca', 'X_umap', 'X_glue'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
atac
AnnData object with n_obs × n_vars = 14566 × 109789
obs: 'balancing_weight'
var: 'chrom', 'chromStart', 'chromEnd', 'highly_variable'
uns: 'neighbors', 'umap', '__scglue__'
obsm: 'X_lsi', 'X_umap', 'X_glue'
obsp: 'distances', 'connectivities'
rna.obs['feature']='rna'
atac.obs['feature']='atac'
combined = ad.concat([rna, atac])
combined
AnnData object with n_obs × n_vars = 29132 × 0
obs: 'balancing_weight', 'feature'
obsm: 'X_umap', 'X_glue'
sc.pp.neighbors(combined, use_rep="X_glue", metric="cosine")
sc.tl.umap(combined)
sc.pl.umap(combined)
sc.tl.leiden(combined,resolution=0.8)
sc.pl.umap(combined,color='leiden')

combined
AnnData object with n_obs × n_vars = 29132 × 0
obs: 'balancing_weight', 'feature', 'leiden'
uns: 'neighbors', 'umap', 'leiden', 'leiden_colors'
obsm: 'X_umap', 'X_glue'
obsp: 'distances', 'connectivities'
combined_rna = combined[combined.obs.feature == 'rna']
combined_rna
View of AnnData object with n_obs × n_vars = 14566 × 0
obs: 'balancing_weight', 'feature', 'leiden'
uns: 'neighbors', 'umap', 'leiden', 'leiden_colors'
obsm: 'X_umap', 'X_glue'
obsp: 'distances', 'connectivities'
combined_rna.obs
| balancing_weight | feature | leiden | |
|---|---|---|---|
| AAACAGCCAGAACCGA-1 | 0.766128 | rna | 0 |
| AAACAGCCAGCCTAAC-1 | 3.160764 | rna | 1 |
| AAACAGCCATATTGAC-1 | 1.073027 | rna | 4 |
| AAACATGCAAATTCGT-1 | 1.073027 | rna | 4 |
| AAACATGCAACCTAAT-1 | 0.660823 | rna | 9 |
| ... | ... | ... | ... |
| TTTGTTGGTATGGTGC-1 | 0.432174 | rna | 12 |
| TTTGTTGGTCAATACG-1 | 1.097787 | rna | 3 |
| TTTGTTGGTCAGGAAG-1 | 0.687277 | rna | 1 |
| TTTGTTGGTTCAAGAT-1 | 1.884496 | rna | 6 |
| TTTGTTGGTTTACTTG-1 | 0.687277 | rna | 1 |
14566 rows × 3 columns
combined_rna.obs.loc[:,'new_leiden'] = np.nan
scripro.glue_metacell(combined_rna,50)
rna_leiden_clusters = combined_rna.obs['new_leiden']
rna_leiden_clusters
AAACAGCCAGAACCGA-1 0_1
AAACAGCCAGCCTAAC-1 1_4
AAACAGCCATATTGAC-1 4_0
AAACATGCAAATTCGT-1 4_0
AAACATGCAACCTAAT-1 9_0
...
TTTGTTGGTATGGTGC-1 12_0
TTTGTTGGTCAATACG-1 3_6
TTTGTTGGTCAGGAAG-1 1_25
TTTGTTGGTTCAAGAT-1 6_0
TTTGTTGGTTTACTTG-1 1_29
Name: new_leiden, Length: 14566, dtype: object
The RNA-Seq and ATAC-seq omics data are combined to generate a new dataset Combined, then divide metacell using the RNA-seq data region, and assign the corresponding metacell to the corresponding ATAC-seq data.
combined_atac = combined[combined.obs.feature == 'atac']
distance_matrix = cdist(combined_atac.obsm['X_umap'], combined_rna.obsm['X_umap'], metric='euclidean')
nearest_rna = np.argmin(distance_matrix, axis=1)
nearest_rna
array([ 1836, 9072, 1954, ..., 13302, 8738, 12567])
atac_leiden_clusters = rna_leiden_clusters[nearest_rna]
atac_leiden_clusters.index = combined_atac.obs.index
rna.obs = combined_rna.obs
cellgroup = pd.DataFrame(atac_leiden_clusters)
cellgroup
| new_leiden | |
|---|---|
| AAACAGCCAGAACCGA-1 | 0_4 |
| AAACAGCCAGCCTAAC-1 | 8_0 |
| AAACAGCCATATTGAC-1 | 15_0 |
| AAACATGCAAATTCGT-1 | 4_1 |
| AAACATGCAACCTAAT-1 | 9_0 |
| ... | ... |
| TTTGTTGGTATGGTGC-1 | 12_0 |
| TTTGTTGGTCAATACG-1 | 1_17 |
| TTTGTTGGTCAGGAAG-1 | 18_0 |
| TTTGTTGGTTCAAGAT-1 | 6_0 |
| TTTGTTGGTTTACTTG-1 | 1_19 |
14566 rows × 1 columns
Calculate metacell and markergene¶
test_data = scripro.Ori_Data(rna,Cell_num=50,use_glue = True)
test_data.get_glue_cluster(rna_leiden_clusters)
test_data.get_positive_marker_gene_parallel()
The data from ATAC-seq is used to generate the corresponding chromatin landscape, that is the bigwig file corresponding to metacell of the same name, which is stored in the folder ‘./bigwig’.
scripro.dataframe_to_sparse_tsv(atac.to_df(), 'test.tsv')
scripro.get_metacell_fragment(cellgroup,'.','./test.tsv',chunksize = 10000000)
scripro.process_tsv('./metacell_fragment/', 'hg38')
share_seq_data = scripro.SCRIPro_Multiome(8,'hg38',test_data)
Calculate the TF activity score¶
share_seq_data.cal_ISD_parallel('./bigwig/')
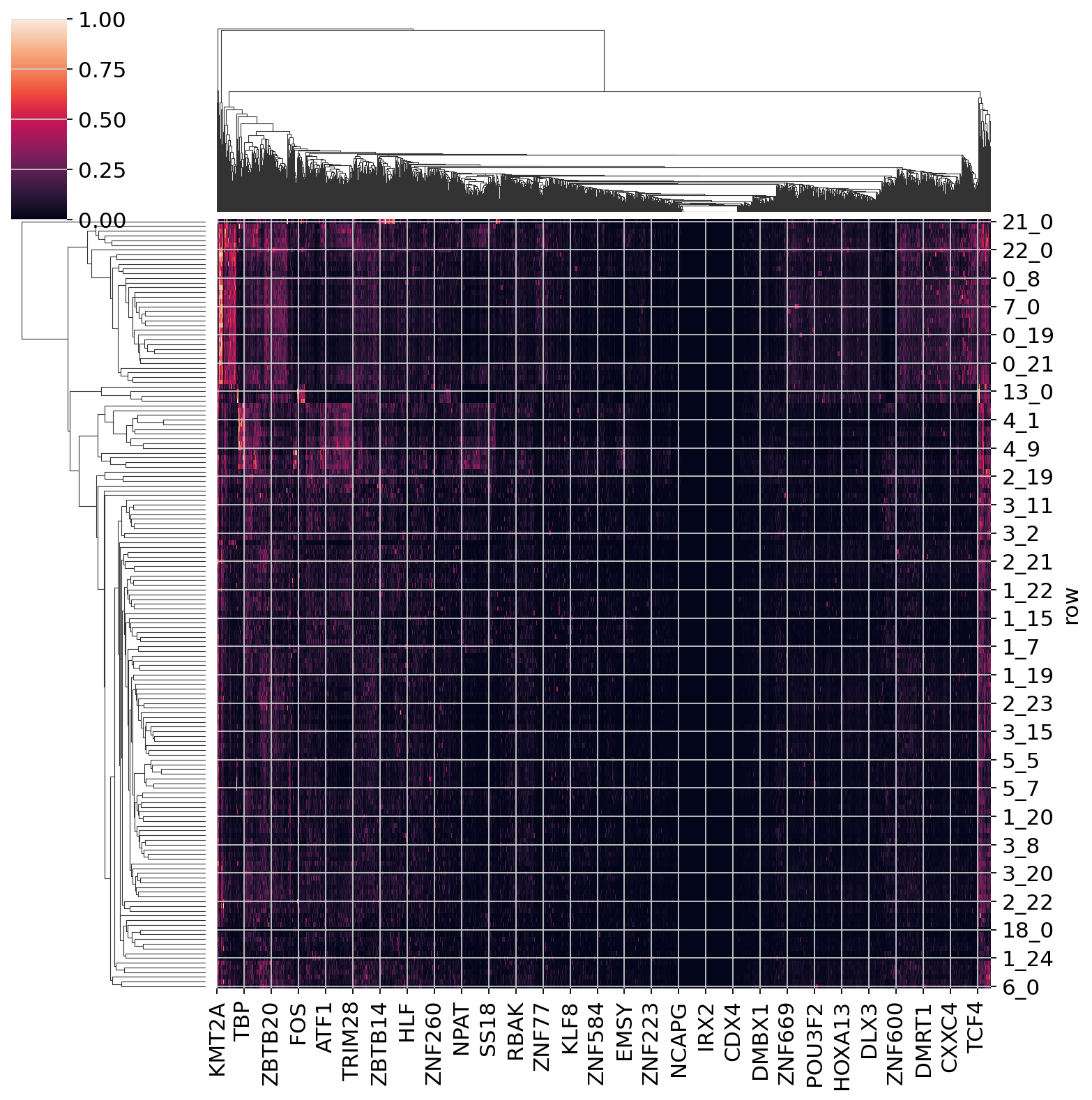
share_seq_data.get_tf_score()
sns.clustermap(share_seq_data.tf_score)